

TINKERtools
@TINKERtoolsMD
Followers
3K
Following
250
Media
55
Statuses
438
The Tinker Molecular Modeling Package: Tinker & Tinker-HP. Developer-Friendly | Free | Polarizable FFs & ML #HPC Tweets: @jppiquem @prenbme J. W. Ponder
Joined January 2017
Start of the Tinker Developers meeting 2025 at @UT_Dallas ! #compchem #biophysics #hpc #supercomputing
1
4
8
#compchem Adaptive sampling (reactive) simulations of the 1M-atom STMV using the FeNNix-Bio1 foundation neural network model coupled to quantum - path integrals- MD. Using a "beyond DFT" version of the model, the PH transition (7 to 5) is studied reaching unprecedented accurary.

(3/3) To bridge the gap between accurate quantum chemistry and condensed-phase Molecular Dynamics, we leverage transfer learning to improve the DFT-based FeNNix-Bio1 foundation model using the DMC/sCI energies and forces. The resulting approach is coupled to path integrals
0
3
16
(3/3) To bridge the gap between accurate quantum chemistry and condensed-phase Molecular Dynamics, we leverage transfer learning to improve the DFT-based FeNNix-Bio1 foundation model using the DMC/sCI energies and forces. The resulting approach is coupled to path integrals
1
4
13
#compchem Second preprint linked to the FeNNix-Bio1 #machinelearning foundation model. FeNNix-Bio1's inference is pretty fast already with a few GPUs but, "what if", we were able to push it at the #Exascale? Let's have a glimpse into the future (1/3): "Pushing the Accuracy Limit
29
35
146
#compchem The FeNNix-Bio1 foundation model's inference is fast and leverages multi-GPUs computing systems (here @nvidia 's H100 nodes). It is also designed so learning a new model remains economical (1 card or node depending on the model size) and can be performed in 48 hours.
#compchem What could we do if we had a fast and accurate foundation #MachineLearning learning model for condensed phase molecular dynamics simulation of biological systems? 👉Check the @ChemRxiv preprint: "A Foundation Model for Accurate Atomistic Simulations in Drug Design":
0
8
29

A Foundation Model for Accurate Atomistic Simulations in Drug Design 1. This paper introduces FeNNix-Bio1, a foundation machine-learning model for atomistic molecular dynamics simulations, aimed at revolutionizing drug design by providing accurate simulations that include
4
17
79

This is exciting stuff. A Foundation Model for Accurate Atomistic Simulations in Drug Design https://t.co/7ZLQszjyLu
0
3
8
AI + physics turns dynamics into a data engine. FeNNix-Bio1: a foundation model that drives QM-level, reactive MD on a single GPU, hydration, folding, binding ΔG within ≈1 kcal/mol, yet pushes 7M-atom boxes at force-field speed
3
18
79
#compchem What could we do if we had a fast and accurate foundation #MachineLearning learning model for condensed phase molecular dynamics simulation of biological systems? 👉Check the @ChemRxiv preprint: "A Foundation Model for Accurate Atomistic Simulations in Drug Design":
1
17
72
#compchem FeNNix-Bio1's full-range of capabilities is demonstrated by modelling: water properties, ions in solution, small molecules hydration free energies, complex folding free-energy landscapes, large-scale protein dynamics, protein-ligand binding and chemical reactions. We
#compchem What could we do if we had a fast and accurate foundation #MachineLearning learning model for condensed phase molecular dynamics simulation of biological systems? 👉Check the @ChemRxiv preprint: "A Foundation Model for Accurate Atomistic Simulations in Drug Design":
0
3
17
FeNNix-Bio1: an Highly scalable foundation #machinelearning model for biosimulations in Tinker-HP/Deep-HP #compchem #HPC
#compchem What could we do if we had a fast and accurate foundation #MachineLearning learning model for condensed phase molecular dynamics simulation of biological systems? 👉Check the @ChemRxiv preprint: "A Foundation Model for Accurate Atomistic Simulations in Drug Design"
0
1
5
#compchem New paper in J. Phys. Chem. Lett @JPhysChem : "Lambda-ABF-OPES: Faster Convergence with High Accuracy in Alchemical Free Energy Calculations". Paper: https://t.co/99ARrN8xk9 preprint: https://t.co/xIv2j1z6Jb To compute absolute binding free energies, this approach
4
19
87
#compchem Just published in JCIM @JCIM_JCTC: "AMOEBA Polarizable Molecular Dynamics Simulations of Guanine Quadruplexes: from the c-Kit Proto-oncogene to HIV-1." Paper: https://t.co/WvyU0IT7VL Updated preprint: https://t.co/ghkfdKp3W1 Very nice work by @ElAhdab_Dina. Another
0
16
101
#compchem Happy to see this one out in @NatureComms: "Histidine 73 methylation coordinates beta-actin plasticity in response to key environmental factors". https://t.co/E4gDlIBzsc We used large scale molecular dynamics simulations coupling adaptive sampling and the AMOEBA
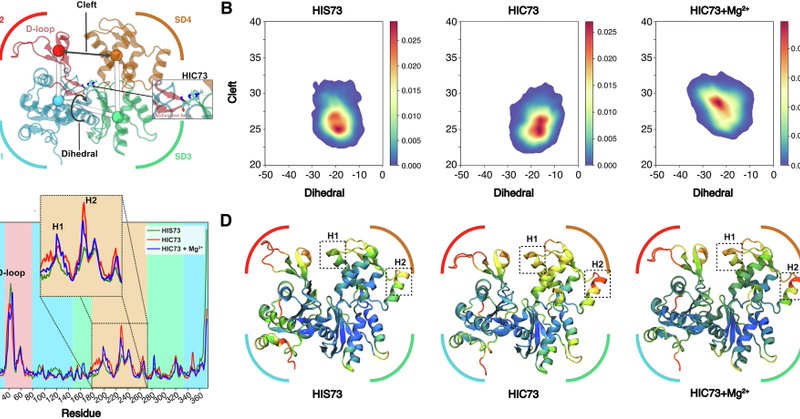
nature.com
Nature Communications - Histidine 73 methylation in β-actin modulates actin plasticity, affecting monomer dynamics and filament stability. Using molecular dynamics simulations, the study...
0
4
36
#compchem New paper in JCTC @JCIM_JCTC : "Velocity Jumps for Molecular Dynamics" https://t.co/vLG6y5zUcu We introduce the Velocity Jumps approach, denoted as JUMP, a new class of Molecular dynamics integrators, replacing the Langevin dynamics by a hybrid model combining a
1
10
39
#compchem New group preprint: "Lambda-ABF-OPES: Faster Convergence with High Accuracy in Alchemical Free Energy Calculations" https://t.co/760aPCKFcy Converged free energy results at a fraction of the cost of standard techniques! Great work by @Narjes_Ansari (@qubit_pharma)
0
15
65
#compchem New preprint: Satellite Tobacco Mosaic Virus: Revealing Environmental Drivers of Capsid and Nucleocapsid Stability using High-Resolution Simulations. https://t.co/4LYV9LrPsW Large scale MD & metadynamics simulations of the complete STMV using the AMOEBA polarizable FF.
1
12
35